| |
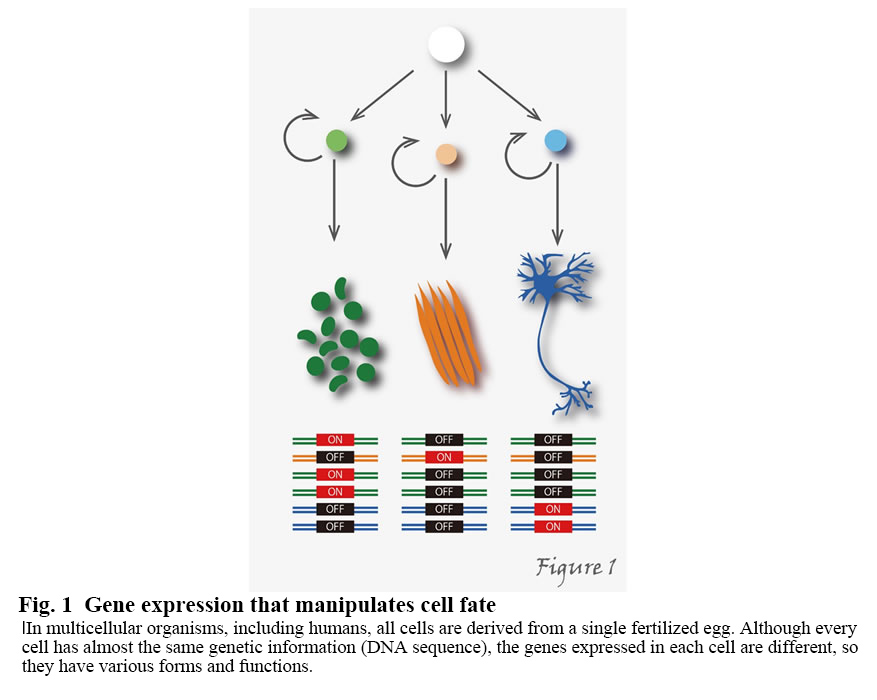
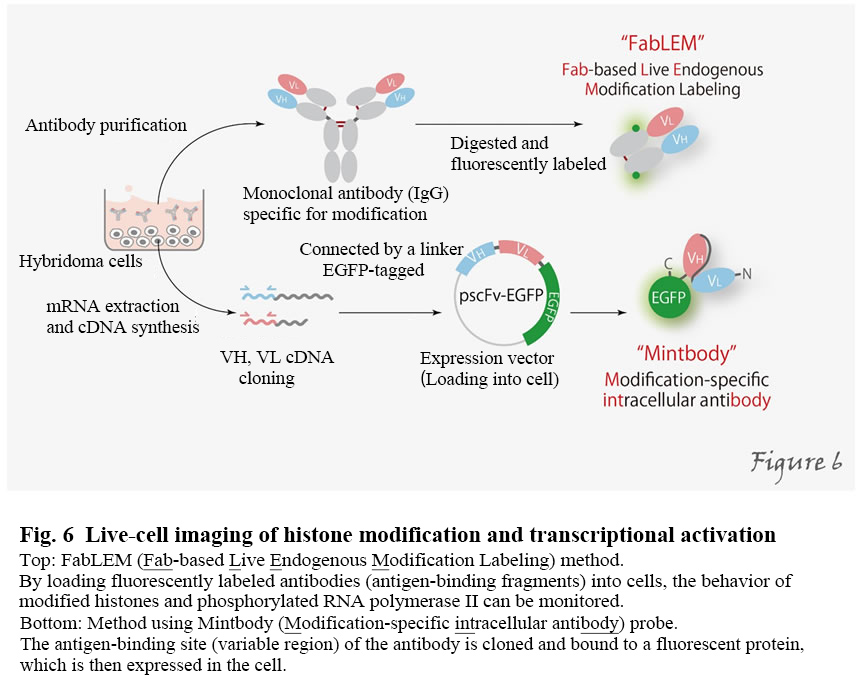
In multicellular organisms, including humans, all cells are derived from a single fertilized egg and so inherently harbor the same genetic information. During development and differentiation, specific gene subsets are expressed in particular cell types. For example, the human body consists of about 60 trillion cells, but why do all the cells have almost the same genetic information (DNA sequence) but different shapes and functions? That's because the genes expressed in individual cells are different (Figure1).
 |
In addition to transcription factors and DNA methylation, post-translational histone modifications, which actively interact with DNA, have been found to play an essential role in the regulation of gene expression. Post-translational histone modifications are known to undergo a wide variety of modifications, particularly those acting to repress or activate transcription. By examining the dynamics of post-translational histone modifications and transcriptional RNA polymerases in living cells, we are attempting to elucidate the mechanisms by which genetic information express in response to cellular functions and external stimuli.
【Background of the research】
What is "histone"?
In eukaryotic cells, genomic DNA is packed into nucleosomes (Figure 2).
The nucleosome consists of 146-147 base pairs DNA wrapped around a histone protein octamer about twice (1.7 times) and is the fundamental unit for storing DNA in the cell nucleus. The histone proteins that work in nucleosome formation call "core" histones; the histone octamer contains two copies of "core" histones (H2A, H2B, H3, and H4). In the nucleus of a cell, there is a very high density of chromatin, which fold nucleosomes, but its distribution is not homogeneous. In general, decondensed chromatin regions are transcriptionally active, and condensed chromatin regions are transcriptionally inactive.
Post-translational histone modifications and epigenetics
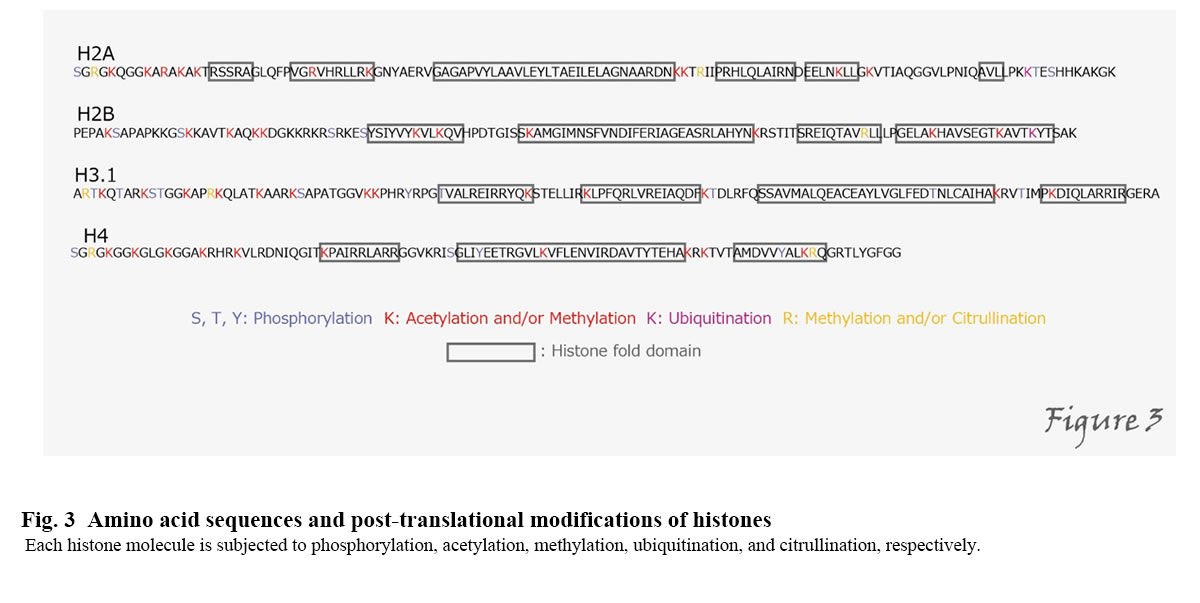
The formation of chromatin structures is not only responsible for DNA storage but also for transcriptional regulation, DNA replication, and damage repair. Post-translational modification of histones plays a particularly important role in their regulation(Kimura, 2013). Histones are subjected to various modifications, including acetylation, methylation, and phosphorylation, on different amino acid residues(Figure3).
 |
For example, acetylation of histone H3 is mainly correlated with transcriptional activation, while methylation of the 9th and 27th lysine residues of H3 is associated with repression of transcription. Gene expression is regulated through these histone modifications, so that the repressed state of transcription, etc., is maintained and inherited even through cell division. Inheritance, such as repression of gene expression without DNA sequence changes, is called "epigenetics." Genomic information involving epigenetic information such as histone modifications and DNA methylation is called "epigenome." The regulation of epigenetics by histone modification is a universal phenomenon found in organisms from yeast to humans. The elucidation of the control mechanism will lead to the development of many technologies using cell engineering, including regenerative medicine, cancer treatment, and stem cell formation.
RNA polymerase
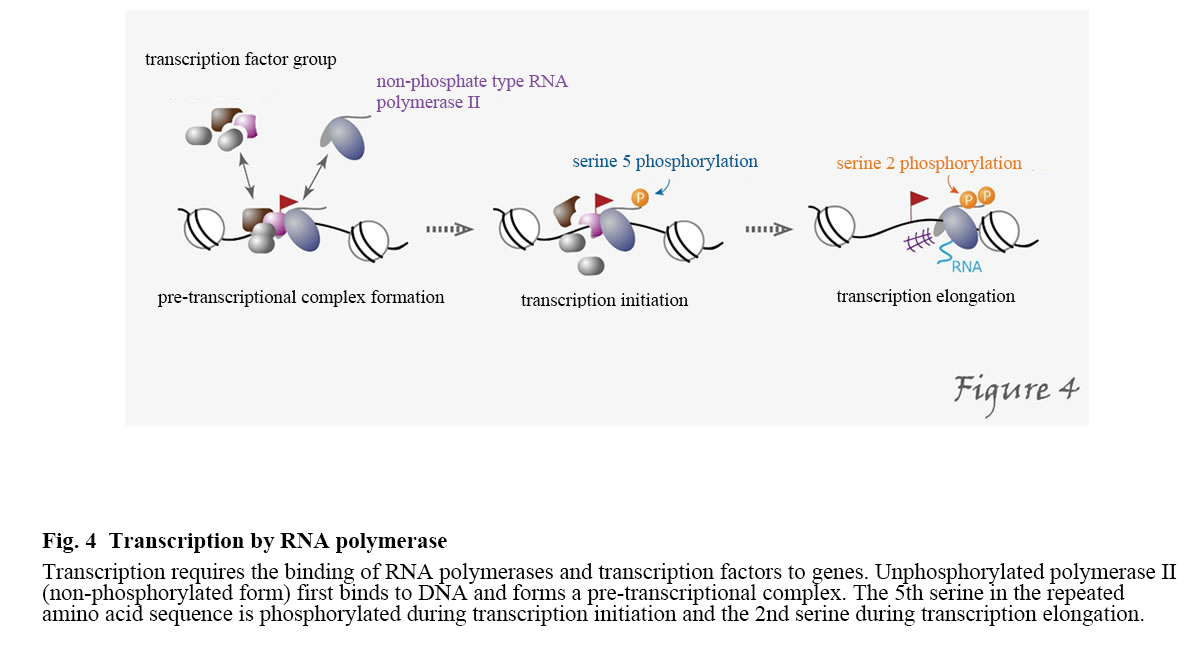
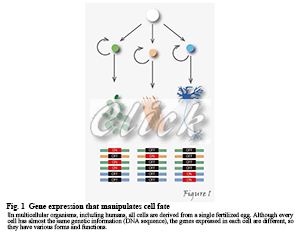
RNA polymerase is a protein that synthesizes RNA (i.e., transcribe it) from a DNA template. In eukaryotic cell nuclei, there are three RNA polymerases: RNA polymerase I is a ribosomal RNA; RNA polymerase II is responsible for the transcription of protein-coding genes and some low-molecular RNAs; RNA polymerase III is responsible for the transcription of low-molecular RNAs such as tRNA. For RNA polymerase II to perform transcription, it first forms a pre-transcriptional complex on the promoter and the essential transcription factor, which leads to the initiation and elongation of transcription. In this process, serine residues in heptad repeats within the C-terminal domain of the largest subunit of RNA polymerase II become phosphorylated. In the C-terminal domain of human RNA polymerase II, the seven amines Tyr-Ser-Pro-Thr-Ser-Pro-Ser repeat 52 times. Still, the fifth serine in the repeated amino acid sequence is phosphorylated during transcription initiation and the second serine during transcription elongation (Figure 4).
 |
Live-cell imaging of histone modification and transcriptional activation
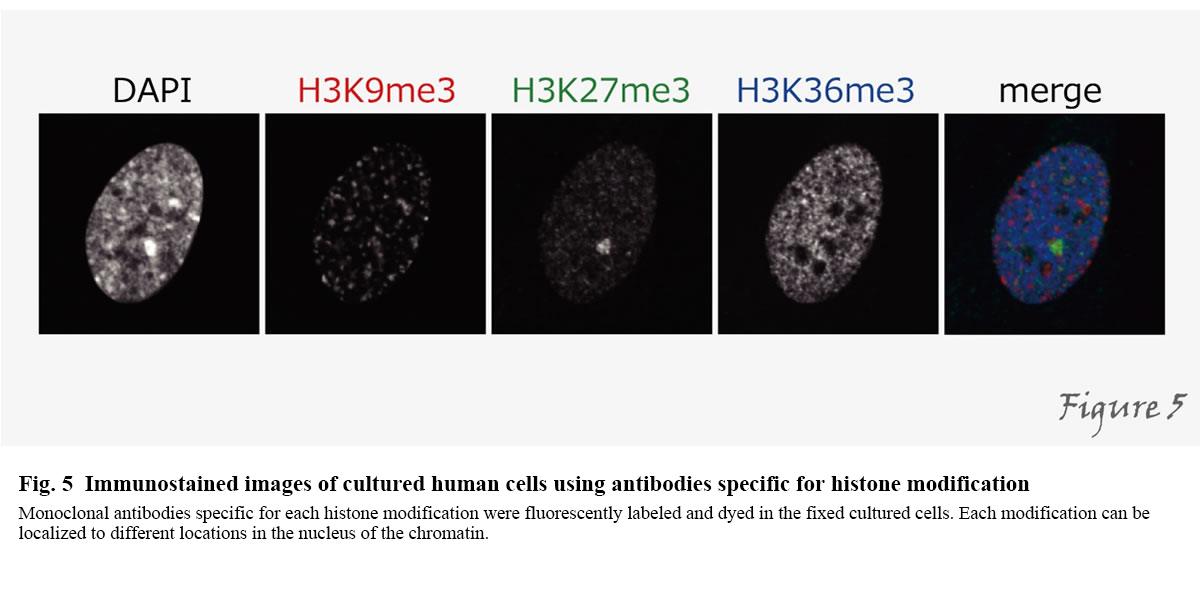
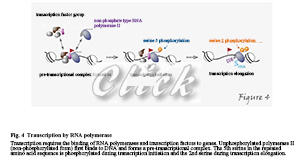
How do the factors we have described so far play an active role in the cell? By fusing with fluorescent proteins, the localization of arbitrary proteins can be observed in living cells. However, not all protein molecules have similar functions, and it is not possible to detect the function of a protein by merely looking at the localization of all proteins. Since many proteins, such as histones and RNA polymerase II, undergo various post-translational modifications depending on their functions, only molecules with special features can be detected using these as indicators. For this purpose, we have developed different specific monoclonal antibodies directed against specific modifications. By using specific antibodies, we can examine the localization of intracellular modifications by immunoprecipitation and immunostaining (Figure 5).
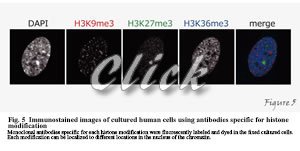 |
However, standard immunostaining methods use fixed and biochemical cells, which cannot follow the dynamic modifications changes. We have successfully developed a method to track modified histones and phosphorylated RNA polymerase II using fluorescently labeled antigen-binding fragments (Fabs), loaded into living cells (Kimura et al. 2010). We named this method FabLEM (Fab-based live endogenous modification labeling) (Hayashi-Takanaka et al. 2011). We have also developed a system that can clone the antigen-binding site (variable region) of antibodies and express fused with fluorescent proteins. (Sato et al. 2013; 2016). This method of selectively visualizing only molecules that have undergone specific post-translational modifications in living cells or in vivo may be beneficial for investigating protein function (Figure 6).
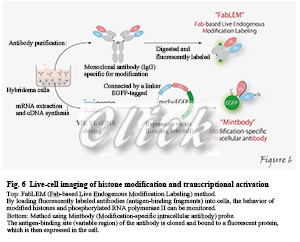 |
【Research content】
Post-translational modifications of histones play a critical role in the regulation of chromosome functions such as gene expression and DNA damage repair. We have generated monoclonal antibodies against various specific modifications to elucidate the significance and regulatory mechanisms of different histone modifications (Kimura et al. 2008). We use these antibodies to investigate the changes in histone modifications throughout the cell cycle, as well as during development, differentiation, and response to external stimuli. Histone modifications can vary in individual gene regions as well as in overall amounts. The distribution of histone modifications across the whole genome can be studied by chromatin immunoprecipitation followed by sequencing (so-called ChIP-seq method) using specific antibodies. We are also developing a system to examine the distribution of histone modifications using small numbers of cells (Harada et al. 2019).
On the other hand, the subcellular localization and global level of histone modifications can be analyzed by immunostaining using fixed and biochemical cells. Still, we have developed methods to investigate the dynamics of histone modifications and transcription in living cells and in vivo. These methods have revealed that the regulation of the phosphorylation of histone H3 involved in chromosome segregation differs between normal and cancerous cells. And that transcriptional regulation is related to histone H3 acetylation response to deacetylase inhibitor (Hayashi-Takanaka et al. 2009; 2011; Sato et al. 2013). We also found that the acetylation of histone H3 facilitates the process from transcriptional initiation to elongation (Stasevich et al. 2014). We are currently investigating dynamic changes of various histone modifications during cell differentiation and in response to external stimuli (Sato et al. 2019).
【References】
Sato Y, Hilbert L, Oda H, Wan Y, Heddleston JM, Chew TL, Zaburdaev V, Keller P, Lionnet T, Vastenhouw N, Kimura H. (2019), an Y, Heddleston JM, Chew TL, Zaburdaev V, Keller P, Lionnet T, Vastenhouw N, Kimura H. Histone H3K27 acetylation precedes active transcription during zebrafish zygotic genome activation as revealed by live-cell analysis. Development 146, dev179127.
Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y, Shirahige K, Kurumizaka H, Kimura H, Ohkawa Y. (2019) A chromatin integration labelling method enables epigenomic profiling with lower input. NCB 21, 287-296.
Sato Y, Kujirai T, Arai R, Asakawa H, Ohtsuki C, Horikoshi N, Yamagata K, Ueda J, Nagase T, Haraguchi T, Hiraoka Y, Kimura A, Kurumizaka H, Kimura H. (2016) A genetically encoded probe for live-cell imaging of H4K20 monomethylation. JMB 428, 3885-3902.
Stasevich TJ, Hayashi-Takanaka Y, Sato Y, Maehara K, Ohkawa Y, Sakata-Sogawa K, Tokunaga M, Nagase T, Nozaki N, McNally JG, and Kimura H. (2014). Regulation of RNA II activation by histone acetylation in single living cells. Nature 516, 272-275.
Sato Y, Mukai M, Ueda J, Muraki M, Stasevich TJ, Horikoshi N, Kujirai T, Kita H, Kimura T, Hira S, Okada Y, Hayashi-Takanaka Y, Obuse C, Kurumizaka H, Kawahara A, Yamagata K, Nozaki N, and Kimura H. (2013). Genetically encoded system to track histone modification in vivo. Sci Rep 3, 2436.
Kimura H. (2013). Histone modification for human epigenome analysis. J Hum Genet 58, 439-445.
Hayashi-Takanaka Y, Yamagata K, Wakayama T, Stasevich TJ, Kainuma T, Tsurimoto T, Tachibana M, Shinkai Y, Kurumizaka H, Nozaki N, and Kimura H. (2011). Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res 39, 6475-6488.
Kimura H, Hayashi-Takanaka Y, and Yamagata K. (2010). Visualization of DNA methylation and histone modifications in living cells. Curr Opin Cell Biol 22, 412-418.
Hayashi-Takanaka Y, Yamagata K, Nozaki N, and Kimura H. (2009). Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol 187, 781-790.
Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, and Nozaki N. (2008). The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct 33, 61-73.

|